Accueil > Pour en savoir plus > Médecine, Santé > Coronavirus > "Un nouveau coronavirus a circulé sans être détecté plusieurs mois avant (…)
"Un nouveau coronavirus a circulé sans être détecté plusieurs mois avant l’apparition des premiers cas de COVID-19 à Wuhan, en Chine : une étude prévoit une émergence dès octobre 2019 ; les simulations indiquent que, dans la plupart des cas, les virus zoonotiques s’éteignent naturellement avant de provoquer une pandémie. Dernières nouvelles" par Jacques Hallard
samedi 3 avril 2021, par
Un nouveau coronavirus a circulé sans être détecté plusieurs mois avant l’apparition des premiers cas de COVID-19 à Wuhan, en Chine : une étude prévoit une émergence dès octobre 2019 ; les simulations indiquent que, dans la plupart des cas, les virus zoonotiques s’éteignent naturellement avant de provoquer une pandémie. Dernières nouvelles
Rapport de l’OMS sur l’origine du virus : Washington et treize pays ’préoccupés’ - AFP, publié le mercredi 31 mars 2021 à 07h11 – Document ‘actu.orange.fr’
Les Etats-Unis et treize pays alliés ont exprimé mardi leurs ’préoccupations’ dans une déclaration commune au sujet du rapport de l’Organisation mondiale de la santé (OMS) sur l’origine du Covid-19, réclamant à la Chine de donner ’pleinement accès’ à ses données.
Le patron de l’OMS, Tedros Adhanom Ghebreyesus, a demandé de son côté une enquête sur l’hypothèse d’une fuite du virus d’un laboratoire en Chine pour expliquer l’origine de la pandémie, et critiqué le partage insuffisant des données par Pékin lors de la mission des experts internationaux cet hiver.
Ces experts, missionnés du 14 janvier au 9 février en Chine, où sont apparus les premiers cas de la maladie en décembre 2019, ont pourtant estimé que l’hypothèse d’une fuite d’un laboratoire était la moins probable…..
Lire l’article complet à la source : https://actu.orange.fr/monde/rapport-de-l-oms-sur-l-origine-du-virus-washington-et-treize-pays-preoccupes-CNT000001yfy5W/photos/le-premier-ministre-britannique-boris-johnson-le-29-mars-2021-a-londres-c2c19c9825893e8d9fdf6bb029b12e53.html
A lire aussi :
Monde - A Wuhan, l’équipe de l’OMS se cantonne à des hypothèses sur l’origine du coronavirus
Orange actu : L’actualité en France et dans le monde en continu

Orange actu : L’actualité en France et dans le monde en continu
Discours attendu du Président Macron ce 31 mars 2021 à 20h00 : les courbes et cartes du Covid en France pour se repérer - Entre fermeture des écoles et confinement strict, mesures départementales ou nationales, le président va devoir trancher, en s’appuyant notamment sur les indicateurs de l’épidémie…. – Par Par Grégory Rozières– 31/03/2021 12:18 CEST - Document ‘huffingtonpost.fr’
Extrait sur la répartition du taux d’incidence par département en France :

Source : Santé publique France • L’incidence représente le nombre de cas pour 100.000 habitants et la positivité le pourcentage de tests positifs. Évolution de l’incidence calculée sur 7 jours
L’article complet sur ce site : https://www.huffingtonpost.fr/entry/discours-de-macron-les-courbes-et-cartes-du-covid-en-france-pour-se-reperer_fr_60642719c5b6fd3650db92e0
Le HuffPost : l’info en accès libre, en article et en vidéo

Journée Nationale du Sport et du Handicap | ANESTAPS
Traduction du 25 mars 2021 par Jacques Hallard – avec ajout de compléments sur la datation moléculaire et les simulations épidémiologiques - de l’article publié le 18 mars 2021 par ‘sciencedaily.com’ sous le titre « Novel coronavirus circulated undetected months before first COVID-19 cases in Wuhan, China » ; accessible sur ce site : https://www.sciencedaily.com/releases/2021/03/210318185328.htm
Source de l’information répercutée : University of California - San Diego ; Etats-Unis
Résumé :
À l’aide d’outils de datation moléculaire et de simulations épidémiologiques, les chercheurs estiment que le virus SRAS-CoV-2 a probablement circulé sans être détecté pendant deux mois, avant que les premiers cas humains de COVID-19 ne soient décrits à Wuhan, en Chine, fin décembre 2019.
Texte complet

Coronavirus illustration (stock image). Credit : © rost9 / stock.adobe.com
À l’aide d’outils de datation moléculaire et de simulations épidémiologiques, des chercheurs de la faculté de médecine de l’Université de Californie à San Diego, avec des collègues de l’Université de l’Arizona et d’’Illumina, Inc.’, estiment que le virus SRAS-CoV-2 circulait probablement sans être détecté pendant au plus deux mois avant que les premiers cas humains de COVID-19 ne soient décrits à Wuhan, en Chine, fin décembre 2019.
Dans le numéro en ligne de la revue ‘Science’ du 18 mars 2021, les auteurs notent également que leurs simulations suggèrent que le virus en mutation s’éteint naturellement dans plus des trois quarts des cas, sans provoquer d’épidémie.
’Notre étude a été conçue pour répondre à la question de savoir combien de temps le SRAS-CoV-2 a pu circuler en Chine avant d’être découvert’, a déclaré l’auteur principal, Joel O. Wertheim , PhD, professeur associé à la division des maladies infectieuses et de la santé publique mondiale de la faculté de médecine de l’UC San Diego.
’Pour répondre à cette question, nous avons combiné trois informations importantes : une compréhension détaillée de la façon dont le SRAS-CoV-2 s’est propagé à Wuhan avant le confinement, la diversité génétique du virus en Chine et les rapports sur les premiers cas de COVID-19 observés en Chine. En combinant ces éléments de preuve disparates, nous avons pu fixer une limite supérieure à la mi-octobre 2019 pour le moment où le SRAS-CoV-2 a commencé à circuler dans la province du Hubei’.
Les cas de COVID-19 ont été signalés pour la première fois fin décembre 2019 à Wuhan, située dans la province du Hubei, dans le centre de la Chine. Le virus s’est rapidement propagé au-delà du Hubei. Les autorités chinoises ont bouclé la région et mis en œuvre des mesures d’atténuation dans tout le pays. En avril 2020, la transmission locale du virus était sous contrôle mais, à ce moment-là, le COVID-19 était devenu pandémique, plus de 100 pays ayant signalé des cas.
Le SRAS-CoV-2 est un coronavirus zoonotique, dont on pense qu’il est passé d’un hôte animal inconnu à l’homme. De nombreux efforts ont été déployés pour déterminer à quel moment le virus a commencé à se propager chez l’homme, sur la base d’enquêtes sur les premiers cas de COVID-19 diagnostiqués. Le premier groupe de cas - et les premiers génomes séquencés du SRAS-CoV-2 - ont été associés au ‘Huanan Seafood Wholesale Market’, mais les auteurs de l’étude affirment qu’il est peu probable que ce groupe de cas ait marqué le début de la pandémie, car les premiers cas documentés de COVID-19 n’avaient aucun lien avec le marché.
Les rapports des journaux régionaux suggèrent que les diagnostics de COVID-19 dans le Hubei remontent au moins au 17 novembre 2019, ce qui suggère que le virus circulait déjà activement lorsque les autorités chinoises ont adopté des mesures de santé publique.
Dans la nouvelle étude, les chercheurs ont utilisé des analyses de l’évolution de l’horloge moléculaire pour tenter de déterminer la date du premier cas, ou cas index, de SRAS-CoV-2. L’expression ’horloge moléculaire’ désigne une technique qui utilise le taux de mutation des gènes pour déduire le moment où deux ou plusieurs formes de vie ont divergé - dans ce cas, le moment où l’ancêtre commun de toutes les variantes du SRAS-CoV-2 a existé, estimé dans cette étude à la mi-novembre 2019.
La datation moléculaire, de l’ancêtre commun le plus récent, est souvent considérée comme synonyme de cas index d’une maladie émergente. Cependant, a déclaré le co-auteur Michael Worobey, PhD, professeur d’écologie et de biologie évolutive à l’Université d’Arizona : ’Le cas index peut être antérieur à l’ancêtre commun - le premier cas réel de cette épidémie peut être survenu quelques jours, semaines ou même plusieurs mois avant l’ancêtre commun estimé. Déterminer la longueur de cette ’fusion phylogénétique’ était au cœur de notre enquête.’
Sur la base de ces travaux, les chercheurs estiment que le nombre médian de personnes infectées par le SRAS-CoV-2 en Chine était inférieur à un jusqu’au 4 novembre 2019. Treize jours plus tard, il était de quatre individus, et de seulement neuf le 1er décembre 2019. Les premières hospitalisations à Wuhan avec une affection identifiée plus tard comme COVID-19, sont survenues à la mi-décembre.
Les auteurs de l’étude ont utilisé une variété d’outils analytiques pour modéliser la façon dont le virus du SRAS-CoV-2 a pu se comporter lors de l’épidémie initiale et des premiers jours de la pandémie, alors qu’il était en grande partie une entité inconnue et que l’ampleur de la menace pour la santé publique n’était pas encore pleinement réalisée.
Ces outils comprenaient des simulations d’épidémies basées sur la biologie connue du virus, comme sa transmissibilité et d’autres facteurs. Dans seulement 29,7 % de ces simulations, le virus était capable de créer des épidémies autonomes. Dans les 70,3 % restants, le virus a infecté relativement peu de personnes avant de s’éteindre. L’épidémie moyenne qui a échoué s’est terminée seulement huit jours après le cas index.
’Habituellement, les scientifiques utilisent la diversité génétique virale pour déterminer le moment où un virus a commencé à se propager’, a déclaré Wertheim. ’Notre étude a ajouté une couche cruciale à cette approche en modélisant combien de temps le virus aurait pu circuler avant de donner lieu à la diversité génétique observée.
’Notre approche a donné des résultats surprenants. Nous avons constaté que plus des deux tiers des épidémies que nous avons tenté de simuler se sont éteintes. Cela signifie que si nous pouvions remonter le temps et répéter l’année 2019 cent fois, deux fois sur trois, le COVID-19 se serait éteint de lui-même sans déclencher de pandémie. Cette constatation corrobore l’idée que les humains sont constamment bombardés d’agents pathogènes zoonotiques.’
Wertheim a noté que même si le SRAS-CoV-2 circulait en Chine à l’automne 2019, le modèle des chercheurs suggère qu’il le faisait à de faibles niveaux jusqu’à au moins décembre de cette année-là.
’Compte tenu de cela, il est difficile de concilier ces faibles niveaux de virus en Chine avec les affirmations d’infections en Europe et aux États-Unis au même moment’, a déclaré Wertheim. ’Je suis assez sceptique quant aux allégations de COVID-19 en dehors de la Chine à cette époque.’
La souche originale du SRAS-CoV-2 est devenue épidémique, écrivent les auteurs, parce qu’elle était largement dispersée, ce qui favorise la persistance, et parce qu’elle s’est développée dans les zones urbaines où la transmission était plus facile. Dans des épidémies simulées impliquant des communautés rurales moins denses, les épidémies se sont éteintes dans 94,5 à 99,6 % des cas.
Depuis, le virus a muté à plusieurs reprises, certains variants devenant plus transmissibles.
’La surveillance des pandémies n’était pas préparée à un virus comme le SRAS-CoV-2’, a déclaré M. Wertheim. ’Nous cherchions le prochain SRAS ou MERS, quelque chose qui tuait des gens à un taux élevé, mais avec le recul, nous voyons comment un virus hautement transmissible avec un taux de mortalité modeste peut aussi faire des ravages dans le monde.’
Les co-auteurs sont : Jonathan Pekar et Niema Moshiri, UC San Diego ; et Konrad Scheffler, Illumina, Inc.
Cette recherche a été financée, en partie, par les ‘National Institutes of Health’ (subventions AI135992, AI136056, T15LM011271), le Google Cloud COVID-19 Research Credits Program, la ‘David and Lucile Packard Foundation’, l’Université d’Arizona et la ‘National Science Foundation’ (subvention 2028040).
Source de l’article : Materials provided by University of California - San Diego. Original written by Scott LaFee. Note : Content may be edited for style and length.
Référence de la publication : Jonathan Pekar, Michael Worobey, Niema Moshiri, Konrad Scheffler, Joel O. Wertheim. Timing the SARS-CoV-2 index case in Hubei province. Science, 2021 ; eabf8003 DOI : 10.1126/science.abf8003
Pour citer cette page : MLA APA Chicago - University of California - San Diego. ’Novel coronavirus circulated undetected months before first COVID-19 cases in Wuhan, China : Study dates emergence to as early as October 2019 ; Simulations suggest in most cases zoonotic viruses die out naturally before causing a pandemic.’ ScienceDaily. ScienceDaily, 18 March 2021. www.sciencedaily.com/releases/2021/03/210318185328.htm.
Articles apparentés :
SARS-CoV-2 Jumped from Bats to Humans Without Much Change, Study Finds
Mar. 12, 2021 — How much did SARS-CoV-2 need to change in order to adapt to its new human host ? New research shows that since December 2019 and for the first 11 months of the SARS-CoV-2 pandemic, there has been very ...
Likely Molecular Mechanisms of SARS-CoV-2 Pathogenesis Are Revealed by Network Biology
Sep. 23, 2020 — Researchers have built an interactome that includes the lung-epithelial cell host interactome integrated with a SARS-CoV-2 interactome. Applying network biology analysis tools to this ...
Potential Therapeutic Agents, Vaccines for COVID-19
Mar. 12, 2020 — Since the first reports of a new coronavirus disease in Wuhan, China, in December 2019, COVID-19 has spread rapidly across the globe, threatening a ...
New Compounds Thwart Multiple Viruses, Including Coronavirus
Feb. 26, 2020 — According to a Feb. 13 report from the World Health Organization, the Wuhan coronavirus has stricken more than 46,000 people and has caused over 1,300 deaths since the first cases in Wuhan, China, in ...
Get the latest science news with ScienceDaily’s free email newsletters, updated daily and weekly. Or view hourly updated newsfeeds in your RSS reader : Email Newsletters RSS Feeds
ScienceDaily : Your source for the latest research news -Contact Us - About This - Site | Staff | Reviews | Contribute | Advertise | Privacy Policy | Editorial Policy | Terms of Use - Copyright 2021 ScienceDaily or by other parties, where indicated. All rights controlled by their respective owners. Content on this website is for information only. It is not intended to provide medical or other professional advice. Views expressed here do not necessarily reflect those of ScienceDaily, its staff, its contributors, or its partners. Financial support for ScienceDaily comes from advertisements and referral programs, where indicated. — CCPA : Do Not Sell My Information — — GDPR : Privacy Settings —

{kind=link}
Retour au début du document traduit
Compléments sur la datation moléculaire
La datation moléculaire – Document ‘evolution-biologique.org/histoire-de-la-vie’

principe de la datation moléculaire
Dans les années soixante, on a déterminé la séquence en acides aminés de l’αhémoglobine humaine puis celle de quelques espèces animales. En 1965, Zucckerland et Pauling ont eu l’idée de les comparer à l’ αhémoglobine humaine. Ils ont constaté que le nombre d’acides aminés différents est d’autant plus élevé que l’espèce est un parent éloigné de l’espèce humaine. Il existe une relation linéaire entre la proportion d’acides aminés substitués et l’âge de l’ancêtre commun espèce / Homme. Cette observation est à la base de la notion d’horloge moléculaire et de la datation moléculaire.
Tout se passe comme si les deux lignées avaient accumulé des mutations régulièrement au cours du temps et indépendamment l’une de l’autre. On disposait donc d’une horloge ayant enregistré le temps écoulé depuis la séparation de l’ancêtre commun.
L’idée vint ensuite d’utiliser cette horloge pour dater l’apparition d’un ancêtre commun sans utiliser les archives fossiles. Imaginons que nous ignorions la date d’apparition de l’ancêtre commun Requin/Homme. Nous connaissons par contre grâce aux archives fossiles les dates d’apparition des ancêtres communs Vache/Homme, Poulet/Homme et Carpe Homme. Nous allons utiliser ces données pour tracer la droite ’proportion d’acides aminés substitués’ en fonction de ’l’âge de l’ancêtre commun’. C’est la phase de calibration de l’horloge biologique.
Une fois l’horloge calibrée nous allons extrapoler la droite en la traçant au delà du point Carpe/Homme. En utilisant la proportion d’acides aminés substitués entre Requin et Homme (0.8), il est possible de déterminer la date d’apparition de l’ancêtre commun (450 millions d’années). Voilà exposé très simplement le principe de la datation moléculaire.
La réalité est moins simple. Depuis les travaux des pionniers, on s’est aperçu que :
- L’horloge ne fonctionnait pas à la même vitesse chez toutes les protéines. Celle du cytochrone C fonctionne 4 fois moins vite que celle de l’hémoglobine. L’hémoglobine n’existant que chez les Vertébrés on ne pourra pas extrapoler le calibrage de son horloge à une autre protéine n’existant par exemple que chez les Mollusques.
- On a également montré que l’horloge d’une même protéine ne fonctionnait pas à la même vitesse dans toutes les lignés descendant d’un même ancêtre commun. Il semble en particulier qu’elle fonctionne plus lentement chez les Vertébrés.
- Les datations paléontologiques sont entachées d’une certaine erreur et évoluent en fonction des découvertes. Par exemple l’interprétation de Kimberella comme proche parent des Mollusques peut amener à recalibrer les horloges des protéines de Mollusques de -530 à -560 millions d’années.
Pour contourner ces difficultés les travaux récents portent sur un grand nombre de protéines prises chez un grand nombre d’êtres vivants appartenant à des lignées aussi différentes que possible. Des programmes informatiques sophistiqués permettent de repérer les décalages dans les vitesses des horloges et de les corriger.
Depuis une dizaine d’années les publications sur le sujet se sont multipliées au rythme des découvertes du génome des êtres vivants.

arbres phylogénétiques des Métazoaires
Les arbres phylogénétiques ci-dessus ont été simplifiés de manière à ne conserver que les noeuds suivants :
1 - ancêtre commun des Opisthochontes (Métazoaires et Champignons)
2 - ancêtre commun des Métazoaires.
3 - ancêtre commun des Bilatéraliens.
A - d’aprés Hedges et al, 2004 (64 protéines, calibration : Vertébrés) ;
B - d’aprés Douzery et al, 2004 (129 proteines, calibration : Végétaux, Champignons, Vertébrés, Arthropodes) ;
C - d’aprés Petersen et al, 2005 (7 proteines, calibration : Insectes, Echinodermes, Mollusques).
La figure ci-dessus montre les grandes variations qui existent entre plusieurs datations moléculaires. En calibrant sur des Vertébrés, la datation A surestime sans doute les dates des différents nœuds. Il existe cependant un hiatus entre datation moléculaire et paléontologie. Avant l’Ediacarien, il n’y a aucun fossile incontestable de Métazoaires. Les Horodyskia, Pararenicola et autres Sinosabellidites trouvés entre -600 millions d’années et - 1500 millions d’années ont tous vu leur statut de Métazoaires remis en cause. Les datations moléculaires sont-elles totalement fausses ? Probablement pas, les Métazoaires ont certainement une histoire avant l’Ediacarien. Peut-être étaient-ils de petite taille et occupaient-ils un milieu qui ne donne pas facilement lieu à la fossilisation. On peut les imaginer pélagiques ou installés près des sources thermales océaniques.
© Copyright 1999-2021 - Ciavatti © Copyright 2004-2021 - CMS Made Simple - Source : http://www.evolution-biologique.org/histoire-de-la-vie/la-vie-pluricellulaire/la-datation-moleculaire.html
Comment les horloges moléculaires donnent l’heure de l’espèce humaine 17 janvier 2018, 22:35 CET • Document ‘theconversation.com’
Il y avait une fois la naissance de l’Homo sapiens… Milan Nykodym/Flickr, CC BY
Cet article est publié dans le cadre de la Nuit Sciences et Lettres : « Les Origines », qui se tiendra le 7 juin 2019 à l’ENS, et dont The Conversation France est partenaire. Retrouvez le programme complet sur le site de l’événement.
L’ADN est le support de notre histoire familiale et plus largement de l’histoire de l’évolution de notre espèce : comment sommes-nous liés à nos parents non-humains chimpanzés ; comment Homo sapiens a rencontré les Néandertaliens ; et comment les gens ont migré hors d’Afrique, s’adaptant à de nouveaux environnements et modes de vie au long de la route. Notre ADN contient également des indices sur le calendrier de ces événements clés dans l’évolution humaine.
Quand les scientifiques disent que les humains modernes ont émergé en Afrique il y a environ 200 000 ans et ont commencé leur expansion mondiale il y a environ 60 000 ans : comment estiment-ils ces dates ?
Traditionnellement, les chercheurs construisaient des chronologies de la préhistoire humaine en se basant sur des fossiles et des artefacts, qui peuvent être directement datés avec des méthodes telles que [ la datation par le Carbone 14 et la datation au potassium-argon. Cependant, ces méthodes nécessitent des vestiges antiques dans certaines conditions de conservation, ce qui n’est pas toujours le cas. De plus, des fossiles ou des artefacts pertinents n’ont pas été découverts pour décrire tous les jalons de l’évolution humaine.
L’analyse de l’ADN des génomes actuels et anciens fournit une approche complémentaire pour la datation des événements de l’évolution. Certains changements génétiques se produisent à un rythme constant par génération, ils fournissent ainsi une estimation du temps qui passe : une horloge moléculaire en somme. En comparant les séquences d’ADN, les généticiens peuvent non seulement reconstituer les relations entre différentes populations ou espèces, mais également déduire l’histoire de l’évolution sur des échelles très grandes.
Grâce à des techniques de séquençage d’ADN plus sophistiqués et à une meilleure compréhension des processus biologiques à l’origine des changements génétiques, les horloges moléculaires deviennent de plus en plus précises. En appliquant ces méthodes à des bases de données ADN toujours plus riches de diverses populations (d’aujourd’hui et du passé), les généticiens aident à construire une chronologie plus précise de l’évolution humaine.
ADN et mutations
Les horloges moléculaires sont basées sur deux processus biologiques clés qui sont la source de toute variation héréditaire : la mutation et la recombinaison.

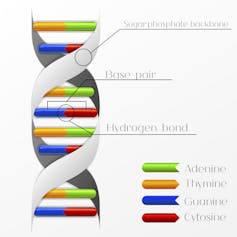{kind=link}
Les mutations sont des changements dans le code de l’ADN, comme quand une base (nucléotide) : A, T, C ou G sont remplacés par une autre par erreur. Shutterstock
Les mutations sont des changements dans les lettres du code génétique de l’ADN – par exemple, un nucléotide guanine (G) devient une thymine (T). Ces changements seront hérités par les générations futures s’ils se produisent dans les cellules œufs issues de la fusion d’un spermatozoïde et d’un ovule. La plupart résultent d’erreurs commises lorsque l’ADN se copie au cours de la division cellulaire, bien que d’autres types de mutations surviennent spontanément ou à la suite de facteurs de risques tels que les radiations et les produits chimiques : des agents mutagènes.
Dans un génome humain, il y a environ 70 changements de nucléotides par génération – une paille quand l’on sait que notre génome est composé de six milliards de lettres. Mais globalement, sur plusieurs générations, ces changements entraînent une variation évolutive substantielle.
Les scientifiques peuvent utiliser les mutations pour estimer le moment où se créent des branches dans notre arbre évolutif, soit la différenciation entre deux espèces. D’abord, ils comparent les séquences d’ADN de deux individus ou espèces, en comptant les différences neutres qui ne modifient pas les chances de survie et de reproduction d’une personne. Puis, connaissant le taux de ces changements, ils peuvent calculer le temps nécessaire pour accumuler autant de différences.
La comparaison de l’ADN entre vous et votre fratrie montrerait relativement peu de différences mutations parce que vous partagiez des ancêtres – maman et papa – il y a seulement une génération. Cependant, il existe des millions de différences entre leshumains et les chimpanzés : notre dernier ancêtre commun aurait vécu il y a plus de six millions d’années.

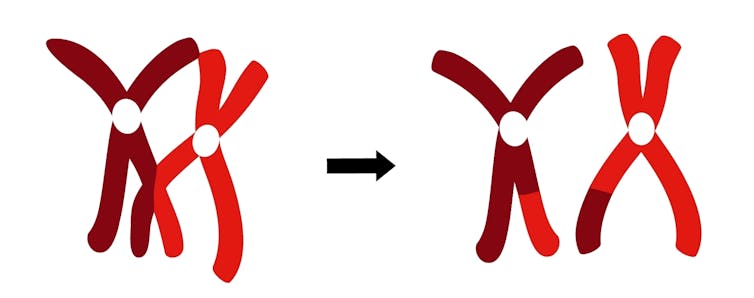{kind=link}
Des fragments des chromosomes de votre mère et de votre père se recombinent avant de vous être transmis. Chromosomes image via Shutterstock.
La recombinaison, également connue sous le nom de crossing-over, est l’autre principale façon avec laquelle l’ADN accumule les changements dans le temps. Il conduit à un brassage des deux copies du génome (une de chaque parent), qui sont regroupées en chromosomes. Au cours de la recombinaison, les chromosomes correspondants (homologues) s’alignent et échangent des segments, de sorte que le génome que vous transmettez à vos enfants est une mosaïque de l’ADN de vos parents.
Chez les humains, environ 36 événements de recombinaison se produisent chaque génération, un ou deux par chromosome. Comme cela se produit à chaque génération, les segments hérités d’un individu particulier se brisent en morceaux de plus en plus petits. Les généticiens peuvent donc se baser sur la taille de ces morceaux et la fréquence des croisements pour estimer à quel moment de l’histoire deux individus ont partagé un ancêtre commun.

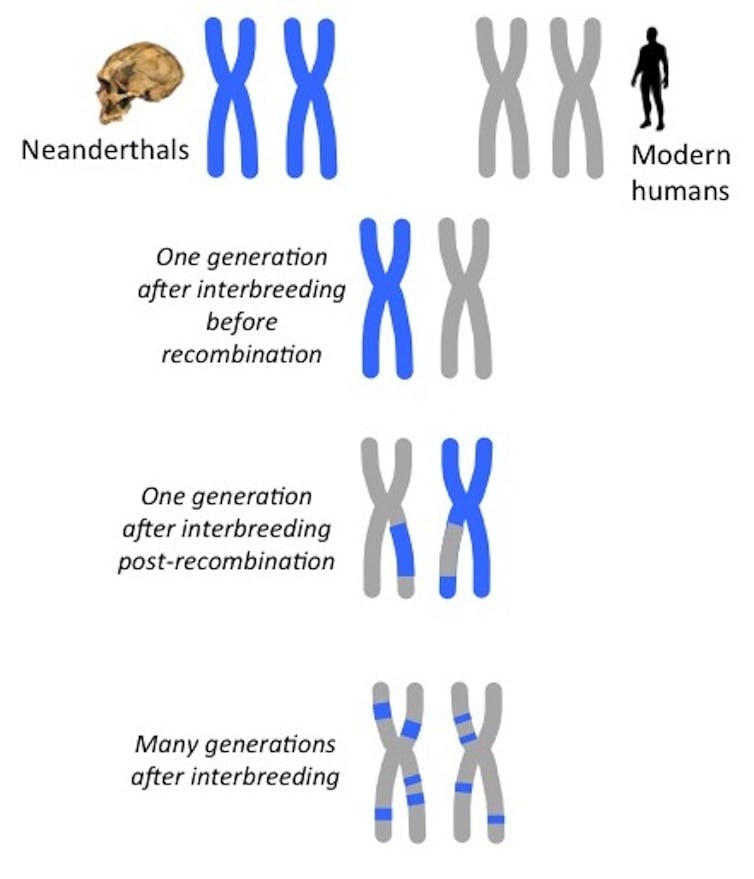{kind=link}
Le flux de gènes entre des populations divergentes conduit à des chromosomes avec une ascendance en mosaïque. Comme la recombinaison se produit à chaque génération, les morceaux d’ascendance néandertalienne dans les génomes humains modernes deviennent de plus en plus petits au fil du temps. Bridget Alex, CC BY-ND
Construire une frise chronologique avec des mutations
Les changements génétiques issus de la mutation et de la recombinaison fournissent deux horloges distinctes, chacune adaptée à la datation de différents événements évolutifs et échelles de temps.
Les mutations s’accumulent très lentement donc cette horloge fonctionne mieux pour les événements très anciens, comme pour les différenciations entre espèces. D’autre part, l’horloge de recombinaison est utile pour dater des évènements ayant eu lieu ces 100 000 dernières années. Ces événements « récents » (en temps évolutif) incluent le flux de gènes entre des populations humaines distinctes, la montée d’adaptations bénéfiques ou l’émergence de maladies génétiques.
Le cas des Néandertaliens illustre bien comment les horloges de mutation et de recombinaison peuvent être utilisées ensemble pour nous aider à démêler des relations ancestrales complexes. Les généticiens estiment qu’il existe entre 1,5 et 2 millions de différences de mutation entre les Néandertaliens et les humains modernes. L’application de l’horloge de mutation à ce compte suggère que les groupes se sont séparés initialement il y a 750 000 à 550 000 ans.
À cette époque, une population – les ancêtres communs des deux groupes humains – s’est séparée géographiquement et génétiquement. Quelques individus du groupe ont migré vers l’Eurasie et au fil du temps ont évolué en Néandertaliens. Ceux qui sont restés en Afrique sont devenus des humains anatomiquement modernes.

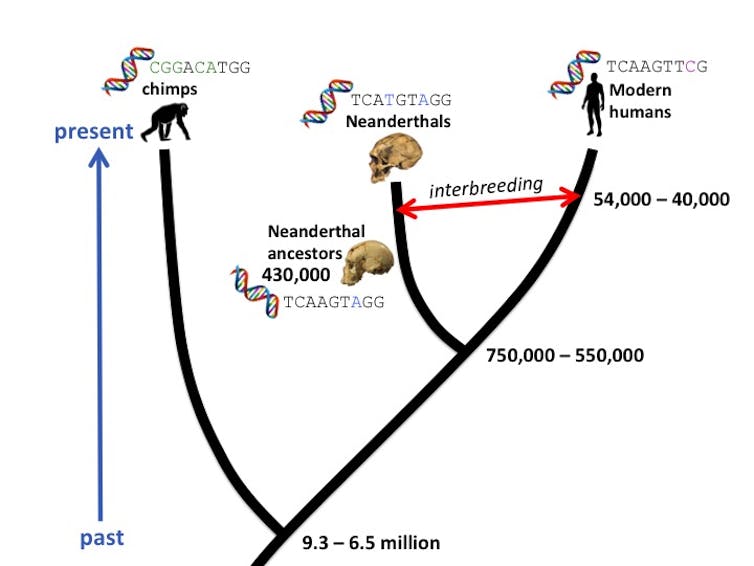{kind=link}
Un arbre phylogénétique affiche les dates de divergence et de croisement que les chercheurs ont estimées avec des méthodes d’horloge moléculaire pour ces groupes. Bridget Alex, CC BY-ND
Cependant, leurs interactions n’étaient pas terminées : les humains modernes ont finalement colonisé l’Eurasie et se sont accouplés avec des Néandertaliens. En appliquant l’horloge de recombinaison à l’ADN néandertalien conservé chez les humains actuels, les chercheurs estiment que les groupes se sont croisés entre 54 000 et 40 000 ans. Lorsque les scientifiques ont analysé un fossile d’Homo sapiens, connu sous le nom d’Oase 1, ayant vécu il y a environ 40 000 ans, ils ont trouvé de grandes régions d’ascendance néandertalienne dans le génome d’Oase 1, suggérant que Oase 1 avait un ancêtre néandertalien il y a seulement quatre ou six générations. En d’autres termes, l’arrière-arrière-grand-parent d’Oase 1 était un néandertalien.

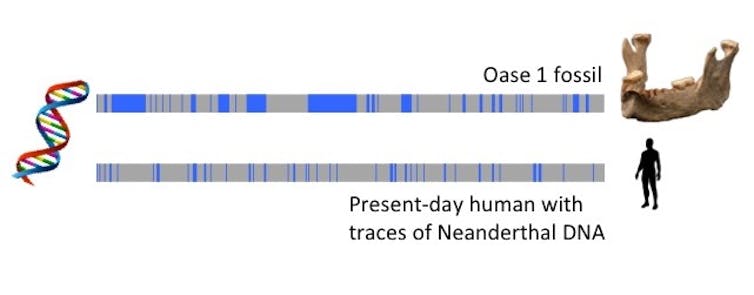{kind=link}
Comparaison du chromosome 6 du fossile Oase vieux de 40 000 ans à un humain d’aujourd’hui. Les bandes bleues représentent des segments d’ADN de Néandertal issus de croisements antérieurs. Les segments d’Oase sont plus longs parce qu’il a eu un ancêtre néandertalien juste 4-6 générations avant qu’il ait vécu, basé sur des estimations utilisant l’horloge de recombinaison. Bridget Alex, CC BY-ND
Des horloges instables
Les horloges moléculaires sont un pilier des calculs évolutifs, non seulement pour les humains mais pour toutes les formes d’organismes vivants. Mais il existe des facteurs rendant complexes les estimations.
Le principal défi provient du fait que les taux de mutation et de recombinaison ne sont pas restés constants au fil de l’évolution humaine. Les taux eux-mêmes évoluent, ils varient donc avec le temps et peuvent différer entre les espèces et même entre les populations humaines. C’est comme si l’on essayait de mesurer le temps avec une horloge qui tourne à différentes vitesses dans différentes conditions.
Un problème concerne un gène appelé Prdm9, qui détermine l’emplacement des cross-overs de l’ADN. Il a été montré que la modification des endroits de l’ADN où ont lieu les recombinaisons est due à la variation de ce gène, et cela chez les humains, les chimpanzés et les souris. En raison de l’évolution de Prdm9, les taux de recombinaison diffèrent entre les humains et les chimpanzés, et peut-être aussi entre les Africains et les Européens. Cela implique que sur des échelles de temps et des populations différentes, l’horloge de recombinaison varie légèrement à mesure que les zones de recombinaisons évoluent.
L’autre problème est que les taux de mutation varient selon le sexe et l’âge. À mesure que les pères vieillissent, ils transmettent quelques mutations supplémentaires à leur progéniture. Le sperme des pères plus âgés a subi plus de cycles de division cellulaire, donc plus de possibilités de mutations. Les mères transmettent moins de mutations (environ 0,25 par an), car les ovules d’une femelle sont formés globalement en même temps, avant sa propre naissance.
Les taux de mutation dépendent également de facteurs tels que le début de la puberté, l’âge à la reproduction et le taux de production de spermatozoïdes. Ces facteurs de vie varient chez les primates vivants et ont probablement aussi différé entre les espèces éteintes des ancêtres humains.
Par conséquent, au cours de l’évolution humaine, le taux de mutation moyen semble avoir considérablement ralenti. Le taux moyen sur des millions d’années depuis la séparation des humains et des chimpanzés a été estimé à environ 1 x10⁻⁹mutations par site et par an – soit environ six lettres d’ADN modifiées par an.
Ce taux est déterminé en divisant le nombre de différences de nucléotides entre les humains et les autres singes par la date de leurs divisions évolutives, déduite à partir de fossiles. Mais lorsque les généticiens mesurent directement les différences nucléotidiques entre parents vivants et enfants, le taux de mutation est moitié moins important : environ 0,5x10⁻⁹par site et par an, soit seulement environ trois mutations par an.
Pour la divergence entre les Néandertaliens et les humains modernes, le taux le plus lent fournit une estimation entre 765 000 à 550 000 ans. Le taux le plus rapide, cependant, suggérerait la moitié de cet âge : 380 000 à 275 000 ans.
Pour savoir quel taux utiliser, les chercheurs ont développé de nouvelles méthodes d’horloges moléculaires, qui répondent aux défis de l’évolution des taux de mutation et de recombinaison.
Une meilleure horloge
Une approche consiste à se concentrer sur les mutations qui surviennent à un rythme constant, indépendamment du sexe, de l’âge et de l’espèce. Cela peut être le cas pour un type particulier de mutation que les généticiens appellent les transitions CpG par lesquelles les nucléotides C deviennent spontanément des T. Les transitions CpG ne résultent généralement pas d’erreurs de copie d’ADN pendant la division cellulaire, leurs taux devraient ainsi être plus uniformes dans le temps.
En se concentrant sur les transitions CpG, les généticiens ont récemment estimé que la fracture entre les humains et les chimpanzés s’était produite entre 9,3 et 6,5 millions d’années, ce qui concorde avec l’âge attendu des fossiles. Même si ces mutations semblent se comporter plus comme une horloge, elles ne sont toujours pas complètement stables.
Une autre approche consiste à développer des modèles qui ajustent les rythmes d’horloge moléculaire en fonction du sexe et d’autres facteurs de la vie. En utilisant cette méthode, les chercheurs ont calculé une divergence chimpanzé-humain compatible avec l’estimation CpG et les dates des fossiles. L’inconvénient ici est que, en ce qui concerne les espèces ancestrales, nous ne pouvons pas être sûrs de certaines caractéristiques, comme l’âge à la puberté ou la durée de vie d’une génération, conduisant à une certaine incertitude dans les estimations.
La solution la plus directe provient des analyses de l’ADN ancien récupéré des fossiles. Les spécimens fossiles sont indépendamment datés par des méthodes géologiques, les généticiens peuvent alors les utiliser pour calibrer les horloges moléculaires pour une période donnée ou une population.
Cette stratégie a récemment résolu le débat sur le moment de notre divergence avec les Néandertaliens. En 2016, des généticiens ont extrait de l’ADN ancien de fossiles de 430 000 ans, ancêtres de Néandertal, après que leur lignée a été séparée de l’Homo sapiens. Sachant où ces fossiles se situent sur l’arbre de l’évolution, les généticiens pourraient confirmer que pour cette période de l’évolution humaine, le taux d’horloge moléculaire plus lent de 0,5 x 10⁻⁹ fournit des dates précises. Cela place la fracture entre 765 000 et 550 000 ans.
À mesure que les généticiens comprennent les subtilités des horloges moléculaires et séquencent toujours plus de génomes, ils sont prêts à comprendre l’évolution humaine, directement à partir de notre ADN.
La version originale de cet article a été publiée en anglais. Tags : génétique évolution Homo sapiens ADN préhistoire
The Conversation : des analyses de l’actualité -Charte de participation Règles de republication Amis de The Conversation Événements Nos flux Faites un don Ce que nous sommes Notre charte Notre équipe Transparence financière Nos membres et partenairesPour les médias Promouvoir The Conversation Contactez-nous - Politique de confidentialité Conditions générales Corrections Mentions légales

Fichier:The Conversation logo.png — Wikipédia
Droits d’auteur © 2010–2021, The Conversation France (assoc. 1901) – Source : https://theconversation.com/comment-les-horloges-moleculaires-donnent-lheure-de-lespece-humaine-89573
Compléments sur les simulations épidémiologiques
La modélisation en pratique dans la gestion d’une épidémie Publié le 12 mai 2020 - Nicole Ladet Rédactrice – Document ‘inrae.fr’
La communication du gouvernement et de nombreux médias ont utilisé des courbes décrivant et quantifiant l’évolution du nombre de décès ou de malades hospitalisés pour expliquer la stratégie de confinement. Elles sont issues soit d’observations permettant de représenter la progression de l’épidémie, soit de simulations permettant d’anticiper l’évolution selon différentes hypothèses. Ces simulations ou prévisions, obtenues par des modèles, permettent d’apprécier les conséquences de telle ou telle décision. Par exemple, qu’est-ce qui se passerait si nous ne faisions rien et laissions la société vivre sa « vie normale » ? Combien de malades, de décès aurions-nous ? Quel effet aurait la fermeture des écoles ou le port généralisé de masques ? A quel moment risquerions-nous d’atteindre la saturation du système de santé ?
illustration - La modélisation en pratique dans la gestion d’une épidémie © Adobe Stock
{kind=link}
A partir de quoi sont réalisées ces prévisions ?
Ces prévisions quantifiées sont des simulations obtenues par des modèles mathématiques qui sont élaborés en plusieurs étapes (Cf. figure 1).

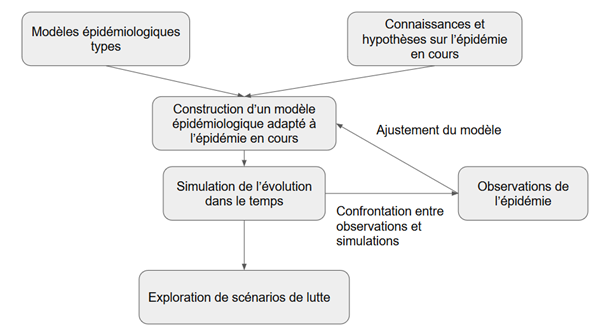{kind=link}
Figure 1. Schéma d’élaboration d’un modèle épidémiologique
Des connaissances et des hypothèses sur l’épidémie servent à élaborer des modèles épidémiologiques. Les prévisions sont ainsi des simulations construites à partir d’hypothèses et des connaissances sur l’épidémie traduites en modèles mathématiques. Les modèles sont ajustés pour que leurs prédictions soient cohérentes avec les données de terrain (nombre de décès, nombre de malades… par exemple). Leur validité est donc étroitement liée à la pertinence des hypothèses sur lesquellesils sont bâtis et à la fiabilité et à la richesse des données disponibles. Les hypothèses sont posées en particulier :
- sur les groupes de populations à considérer. Par exemple, on prend en compte plusieurs classes d’âge car on fait l’hypothèse que la transmission dépend de l’âge.
- sur les choix adoptés pour traduire les connaissances en équation. Par exemple, en l’absence de nouvelles contaminations, on fait l’hypothèse que le nombre de personnes malades décroît exponentiellement.
L’utilisation des données de terrain permet au fil de l’épidémie de ré-évaluer régulièrement la pertinence des modèles et de leurs hypothèses, de les ajuster si nécessaire, et de prévoir de manière quantitative quel serait l’impact de différentes mesures.
Les calculs permettant d’obtenir ces simulations comportent toujours un degré d’incertitude dont il faut tenir compte : chaque résultat associé à un scénario donné est situé dans une fourchette qui s’étend globalement de la prévision la plus pessimiste à la plus optimiste.
Un modèle épidémiologique pour les maladies transmissibles, comment ça marche ?
Le modèle de base épidémiologique considère le nombre de personnes infectées mais aussi le nombre de personnes pas encore atteintes (sensibles) et le nombre de personnes guéries (résistantes) : leur nombre progresse selon la contagiosité de la maladie et selon la part de la population encore sensible, ainsi que celle devenue résistante. On parle de modèle SIR (sensibles, infectés, résistants ou guéris) (Cf. Figure 2).

{kind=link}
Figure 2. Représentation schématique d’un modèle de base SIR avec ses trois compartiments et la définition du taux de reproduction de base R0 associé.
Le taux de reproduction de base R0 représente la contagiosité de l’épidémie : combien de personnes sont infectées à partir d’une personne infectée unique ; il est directement lié au taux de contact et au taux de guérison.
Ce modèle est adapté pour les maladies transmissibles touchant des populations animales aussi bien qu’humaines.

Comprendre une épidémie : cliquez pour en savoir plus
A quoi sert la modélisation ?
Représenter la réalité pour comprendre
Dans un premier temps, un modèle sert à représenter la réalité sous un format simplifié pour comprendre les principaux mécanismes en jeu.
Dans le cas de COVID-19, différentes sources de données concernant la surveillance de l’épidémie à différentes échelles peuvent être utilisées, par exemple :
- les données mondiales disponibles sur le site de Johns Hopkins University (nombre de cas détectés par PCR, nombre de morts)
- des données nationales plus détaillées comme les données hospitalières françaises .
Les modèles peuvent être complexifiés pour mieux représenter la réalité en introduisant par exemple plusieurs catégories de personnes sensibles suivant l’âge, différents symptômes, des populations ayant des comportements différents, ou une dimension spatiale. C’est ce qui a été le cas de la modélisation du COVID-19 avec deux approches :
- par des modèles mathématiques de plus en plus complexes :
- en ajoutant des compartiments pour représenter différentes catégories de personnes (e.g.. le modèle de l’équipe de S. Alizon de l’UMR MIVEGEC qui comporte 10 compartiments) ;
- en prenant en compte l’aléa c’est à dire l’incertitude autour de la survenue d’événements (e.g., modèle de l’équipe de N. Ferguson de l’Imperial College, de l’équipe de V. Colizza de l’iPLESP) ;
- en ajoutant l’espace : la prise en compte des distances, des réseaux d’échange, de l’hétérogénéité dans les territoires.
- par l’ajout d’une approche statistique selon laquelle la probabilité de chaque événement (par exemple être contaminé) est estimée à partir de facteurs comme l’âge et le sexe (e.g., modèle de l’équipe de S Cauchemez). Par ces estimations, on peut affiner la quantification de certains paramètres comme le taux de mortalité selon l’âge et le sexe.
A titre d’illustration, nous représentons un modèle à 11 compartiments développé et calibré au sein de l’UMR EPIA à partir des données françaises (hospitalières et d’autres sources) ainsi que des simulations correspondantes (Cf. Figure 3).

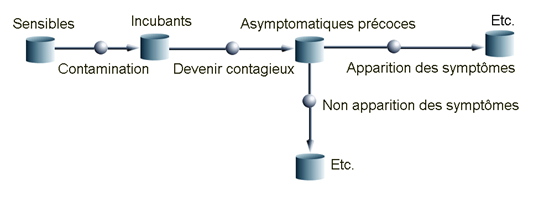{kind=link}
Figure 3a. Extrait du modèle et évolutions temporelles observée et simulée du nombre quotidien de patients en hospitalisation et en service de réanimation en France.
Un extrait du modèle représentant les compartiments initiaux.

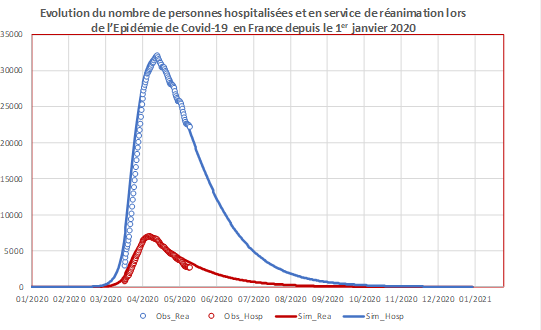{kind=link}
Figure 3b. Extrait du modèle et évolutions temporelles observée et simulée du nombre quotidien de patients en hospitalisation et en service de réanimation en France.
Les points circulaires correspondent aux données hospitalières observées en France jusqu’au 11 mai 2020 (hospitalisations en bleu, services de réanimation en rouge).
Les courbes correspondent aux données simulées par un modèle développé par l’UMR EPIA (hospitalisation en bleu, service de réanimation en rouge) à partir d’un modèle compartimental simple sans dimension spatiale avec prise en compte du confinement à partir du 17 mars 2020 et calibré sur les données disponibles sur le site de Johns Hopkins University et sur le site de data.gouv.fr sur les données hospitalières français. Il n’est pas envisagé dans cet exemple de lever de confinement.
La collaboration de tous est nécessaire
Dans le cas d’une maladie émergente sur laquelle la connaissance est en train de se construire, ces prévisions doivent s’affiner à mesure que les connaissances se renforcent. La collaboration des scientifiques mais aussi de toutes les parties prenantes de l’épidémie est ici essentielle pour mettre à disposition les données et les connaissances : médecins, systèmes de surveillance, gestionnaires de santé, industries de la santé. La gestion d’une épidémie implique pour les décideurs de prendre en compte les aspects sanitaires, y compris la prise en charge des besoins de santé sans lien avec l’épidémie, mais aussi les conséquences socio-économiques générées par la fermeture des écoles, le confinement des travailleurs, la modification des chaînes d’approvisionnement, le recueil de données personnelles.
Explorer des scénarios
Face à une nouvelle épidémie, il faut définir une stratégie de lutte, explorer si on veut viser l’atténuation de l’épidémie ou la suppression. La modélisation permet à moindre coût d’explorer des scénarios intermédiaires basés sur l’utilisation de différents leviers pour limiter la transmission (modalités du lever du confinement, capacité à identifier et isoler les personnes infectantes, traitement réduisant le taux de mortalité ou le taux de passage en réanimation…) en attendant l’arrivée à plus long terme d’un vaccin efficace. Elle est essentielle pour anticiper les conséquences de ces différents scénarios car elle est la base de l’expertise sanitaire en appui aux politiques publiques.
Anticiper le futur et quantifier les effets des choix de politiques de santé publique
Les modèles sont particulièrement utiles actuellement pour explorer les scénarios de déconfinement avec différents leviers possibles pour limiter la transmission. Pendant le confinement en France métropolitaine, le R0 a diminué d’environ 3,4-3,3 avant confinement à une valeur proche de 0.7-0,5, soit une baisse d’environ 85 % (cf. référence).L’objectif est de comprendre les conditions dans lesquelles la maladie est maîtrisée grâce à une transmission du virus limitée (R0 < 1) ou faible (autour de 1) afin de limiter les conséquences sanitaires de la circulation du virus, tout en réduisant les conséquences socio-économiques du confinement.
On peut schématiquement représenter les différentes mesures qui s’offrent à nous pour contrôler la maladie : mesures sanitaires, mesures médicales et vaccination (Cf. figure 4) .

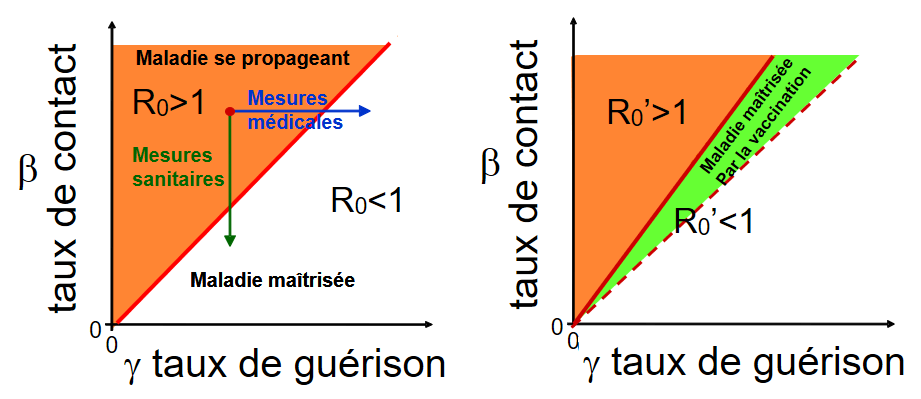{kind=link}
Figure 4 - Représentation du R0 = taux de contact / taux de guérison .
Schéma de gauche : La droite correspond au cas où R0 = 1, c’est à dire où le taux de contact est égal au taux de guérison. Les points au-dessus de la bissectrice (zone en rouge) représentent les cas où le R0>1 et la maladie se propage. Les points en dessous représentent les cas où R0<1 et la maladie est contrôlée. Les mesures médicales tendent à augmenter le taux de guérison et les mesures sanitaires tendent à diminuer le taux de contact.
Schéma de droite : Si on vaccine, la maladie ne se propage que dans la partie non vaccinée de la population et dans ce cas le nouveau R0’ est égal à R0 que multiplie la fréquence de population non vaccinée. La zone où la maladie est maîtrisée après vaccination R0’<1 est plus grande car il y a une nouvelle zone où la maladie est maîtrisée par la vaccination (zone en vert).
Il est intéressant d’associer différentes mesures pour atteindre plus rapidement les conditions pour lesquelles R0<1 ou R0’<1.
Par exemple pour le covid-19, des mesures sanitaires permettent de :
- Limiter le nombre de contacts entre personne infectées et personnes sensibles : confinement régionalisé, organisation du travail (télétravail..), maintien ou pas de la fermeture des écoles, ouvertures partielles des commerces, limitation de rassemblements, tests virologiques pour identifier les personnes infectées et isoler ces personnes et les contacts qu’elles auraient eus durant la période pendant laquelle ils sont porteurs du virus, etc.
- Limiter l’efficacité de la transmission du virus au cours des contacts : mesures barrières (port du masque, lavage des mains, etc.).
Des mesures médicales comme un traitement, permettraient de limiter la durée infectieuse c’est à dire augmenter le taux de guérison. Par ailleurs, l’augmentation du taux de guérison a un autre effet bénéfique en diminuant la mortalité. La vaccination permettrait de limiter le nombre d’individus sensibles, ce qui diminue de fait le R0.
A titre d’illustration, nous représentons avec le modèle précédent l’évolution du nombre de décès en fonction de plusieurs scénarios correspondant à des niveaux de confinement plus ou moins restrictifs (Cf. Figure 5).

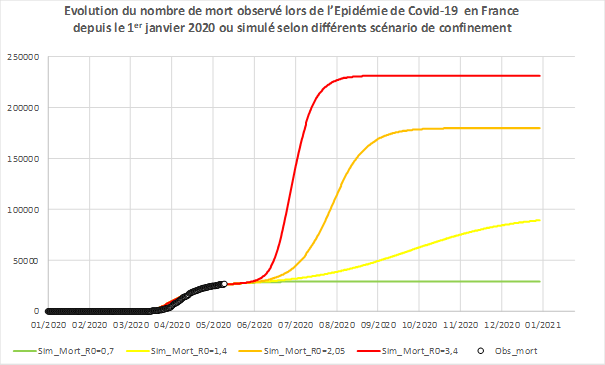{kind=link}
Figure 5 - Evolution temporelle observée et simulée du nombre cumulé de décès quotidien en France. Les points circulaires en noir correspondent aux données de mortalité observées en France jusqu’au 11 mai 2020.
Les courbes correspondent aux données simulées par le même modèle que celui de la figure 3.
- En vert, le confinement s’applique sur toute l’année 2020 (le R0 reste à 0,7).
- Pour les autres courbes, le confinement est levé au 15 mai 2020 mais la transmission est plus ou moins contenue avec diverses actions (port de masques, limitation des regroupements etc).
_ . En jaune, la contagiosité serait le double de celle observée en confinement actuel (R0 = 1,4).
_ . En orange, elle serait à mi-chemin entre celle d’avant confinement et celle durant le confinement (R0=2,05).
_ . En rouge, le confinement est entièrement levé et la transmission reprend avec la même efficacité qu’avant le confinement (pas de geste barrière, etc) (R0 = 3,4).
Ces courbes permettent à chacun de mieux visualiser les raisons qui ont motivé les options de confinement, en visualisant notamment le total des décès épargnés par cette décision (sur le schéma : différence entre la courbe rouge et la courbe verte).
En l’absence de solutions médicales pour diminuer les décès dûes à la maladie et sous les hypothèses envisagées (par exemple, absence de saisonnalité de l’infection), les scénarios jaune orange et rouge avec une contagiosité qui repasserait au-dessus de 1 suite au déconfinement (R0>1) montrent que l’épidémie reprendrait et que les décès recommenceraient à augmenter au bout d‘un certain temps. Cela justifie la mise en place de mesures accompagnant le déconfinement et visant à limiter la contagiosité (tester, tracer, isoler) et pourrait justifier de nouvelles mesures pour contenir l’épidémie, c’est-à-dire infléchir encore la courbe vers le bas pour minimiser le nombre de nouveaux décès liés au COVID-19.
Faire bon usage des prévisions…
Quels que soient leurs niveaux de complexité, les modèles ne sont que des approximations de la réalité, basés sur des hypothèses qui peuvent elles-mêmes évoluer. Par ailleurs, la connaissance de la situation par le grand public modifie les comportements et ainsi la transmission, même en l’absence d’action de santé publique. Aussi, dans le cas d’une nouvelle maladie à répartition mondiale, il est difficile de prédire l’évolution de l’épidémie sur plusieurs mois. Les modèles restent indispensables pour envisager l’avenir et devront être adaptés à l’évolution du contexte et des connaissances comme cela a déjà été le cas en ce début d’année 2020.
Concernant l’épidémie de covid-19, des inconnues cruciales sur la dynamique du virus demeurent. Par exemple, nous savons encore peu de choses sur la force protectrice de la réponse immunitaire contre de futures ré-infections. Nous ne savons pas non plus si la circulation du virus peut être atténuée par les saisons chaudes. Enfin, nous nous ne savons pas si des foyers viraux perdureront au niveau mondial comme c’est le cas de la grippe. Aussi, il est difficile de savoir pendant combien de temps il faudra agir pour limiter la transmission du virus.
Ainsi, les éléments qui permettraient un relâchement total des mesures limitant la transmission seraient qu’ :
- un traitement efficace ait été trouvé, limitant les conséquences et la propagation de l’épidémie ;
- un vaccin efficace ait été mis au point, au mieux d’ici 18 mois à 2 ans, permettant de mettre fin à la circulation du virus sans nouvelles infections ;
- la population ait acquis suffisamment d’immunité collective (60%-70%) pour limiter la transmission dans un fonctionnement normal de notre société. Mais cela représente des morts supplémentaires le temps que l’immunité s’installe.
Si on arrivait à supprimer la circulation du virus en France ou en Europe du fait de mesures sanitaires, donc sans acquisition d’une immunité de groupe, il faudrait veiller à limiter l’introduction de nouveaux cas par
- les mesures efficaces de surveillance sur les réintroductions de la maladie
- ou une réduction drastique de la circulation au niveau mondial.
Etant donné le peu de visibilité que nous avons sur ces différents éléments, il est important d’agir pour limiter la transmission du virus dans les mois qui viennent, après le déconfinement partiel, et d’acquérir des informations pour mieux préciser l’avenir.
PCR : Polymerase Chain Reaction
Références :
a/ Henrik Salje, Cécile Tran Kiem, Noémie Lefrancq, Noémie Courtejoie, Paolo Bosetti, et al.. Estimating the burden of SARS-CoV-2 in France. 2020. pasteur-02548181
b/ Lionel Roques, Etienne Klein, Julien Papaix, Antoine Sar, Samuel Soubeyrand.2020. Effect of a one-month lockdown on the epidemic dynamics of COVID-19 in France. https://hal.archives-ouvertes.fr/hal-02550441 INRAE BioSP - Unité de Biostatistiques et Processus Spatiaux
Voir nos actus sur COVID-19- _AccueilSiège INRAE : 147 rue de l’Université 75338 Paris Cedex 07 - tél. : +33(0)1 42 75 90 00 - Copyright - ©INRAE - CréditsMentions légalesAchatsAccessibilité : non conformeContact

Fichier:Logo-INRAE Transparent.svg — Wikipédia
Source : https://www.inrae.fr/actualites/modelisation-pratique-gestion-dune-epidemie
Analyse - Un an de Covid-19 : les modélisations des scientifiques ne se sont quasiment jamais trompées Par Olivier Monod publié le 17 mars 2021 à 7h28 - Document ‘liberation.fr > Article complet réservé aux abonnés
Alors que le Conseil scientifique préconise plus de mesures de restriction, le journal « Libération » s’est plongé dans un an de projections, de modélisations et d’avis. Le bilan penche largement en faveur des épidémiologistes.

https://www.liberation.fr/resizer/bwsIBmKfr3zuS5WghzsWMDFZTAY=/800x0/filters:format(png):quality(70)/cloudfront-eu-central-1.images.arcpublishing.com/liberation/N3MHIHGH3RH5THR4CXVRQRT6VY.png
« Quand je relis le premier avis du Conseil scientifique, je ne vois pas grand-chose à modifier aujourd’hui ». Simon Cauchemez, épidémiologiste de l’Institut Pasteur, est en charge des modélisations qui nourrissent le Conseil scientifique. Un an après le début de la crise sanitaire, il se livre à l’exercice du retour d’expérience. Pour lui, le rôle premier des modèles n’est pas tant de se projeter que d’analyser les données présentes. « Depuis le début de la crise, nos estimations du taux de reproduction ou de la sévérité du Covid n’ont pas trop évolué. C’est le degré d’incertitude qui s’est réduit », détaille-t-il.
Au début de la pandémie, les simulations épidémiques ont été très écoutées par le pouvoir. Quand Emmanuel Macron annonce le premier confinement en France, le 16 mars 2020, le pays déplore seulement 149 morts. Mais les hôpitaux de trois régions saturent et un modèle de l’’Imperial College’ de Londres prédit entre 300.000 et 500.000 morts en France si l’épidémie suit …
Il vous reste 85% de l’article à lire. Libération réserve cet article à ses abonnés. Vous êtes abonné ? Connectez-vous- Pour lire la suite, abonnez-vous 1 mois pour 1€ Sans engagement S’abonner pour lire l’article
Retour au début du document traduit
Traduction, compléments et intégration de liens hypertextes par Jacques HALLARD, Ingénieur CNAM, consultant indépendant – 31/O3/2020
Site ISIAS = Introduire les Sciences et les Intégrer dans des Alternatives Sociétales
Adresse : 585 Chemin du Malpas 13940 Mollégès France
Courriel : jacques.hallard921@orange.fr
Fichier : ISIAS Coronavirus Novel coronavirus circulated undetected months before first COVID-19 cases in Wuhan, China.4
Mis en ligne par le co-rédacteur Pascal Paquin du site inter-associatif, coopératif, gratuit, sans publicité, indépendant de tout parti, géré par Yonne Lautre : https://yonnelautre.fr - Pour s’inscrire à nos lettres d’info > https://yonnelautre.fr/spip.php?breve103

http://yonnelautre.fr/local/cache-vignettes/L160xH109/arton1769-a3646.jpg?1510324931
{kind=link}
— -